Angioedema - Kaplan A (Updated 2019)
Angioedema
Updated: July 2019
Updated: February 2014
Originally posted: September 2004

Allen P. Kaplan, MD
Medical University of South Carolina
Dept. of Medicine: Pulmonary 96 Jonathan Lucas Street
Charleston, SC 29425
USA
Definition
Angioedema is a transient swelling of the skin or submucosal surface due to increased vascular permeability of small venules. The overlying skin may be normal or mildly erythematous.
Symptoms
The swelling may or may not be associated with urticaria (hives). The onset of angioedema may be associated with a tingling or numb sensation followed (typically) by visible swelling over a period of hours. Itch is minimal compared to that of urticaria. A burning-like dysesthesia may be seen, particularly if caused by bradykinin. Common areas of involvement are hands, feet, face (lips, cheeks, periorbital), and genitalia. There may be swelling of the tongue, pharynx, or larynx. Any of the latter can effect speech and swallowing. Laryngeal edema (vocal cords, glottis) leads to hoarseness and in its most severe form, stridor (difficulty inhaling thru a narrowed opening) and asphyxia.
Classification
It is useful to separate angioedema that is clearly associated with urticaria from angioedema without urticaria. It can also be acquired or hereditary. Hereditary forms of angioedema described thus far occur in the absence of urticaria.
Causes
Angioedema with urticaria
When urticaria and angioedema are present concomitantly, the cause can be considered to be the result of cutaneous mast cell degranulation with release of histamine, leucotrienes, cytokines, and chemokines. Their source can be the aforementioned mast cells, endothelial cells, or blood cells (lymphocytes, monocytes, neutrophils, eosinophils, basophils) that have migrated to the site.
Common allergic causes of angioedema are food or drug reactions due to IgE antibody. It is important to emphasize that although urticaria accompanies angioedema in most instances, angioedema can be seen without urticaria yet the cause and pathogenic mechanism(s) are the same. The depth of the reaction within the skin does differ: urticaria involves blood vessels in the superficial dermis while angioedema involves blood vessel in the deep dermis and subcutaneous tissue. Additional allergens that can cause angioedema are venoms from bee, wasp, yellow jacket, hornet, or fire ants; here too there is IgE antibody to the allergen bound to the surface of cutaneous mast cells via the IgE receptor. Once allergen enters the skin via GI absorption (food, drugs) or injection (drugs, venoms), there is cross-linking of IgE by the allergen, signal transduction thru the IgE receptor, and mast cell activation and secretion.
The same process can occur in reactions that are non-allergic, i.e. not caused by the production of IgE antibody. Exposure to radiocontrast medium is an example in which changes in the osmolal environment of the mast cell leads to degranulation. Morphine and other opiate-derived medications can cause mast cell degranulation via stimulation of opiate receptors and angioedema can be one of the manifestations. This is also true of adverse reactions to NSAIDS (non-steroidal anti-inflammatory drugs) which alter the metabolism of arachidonic acid (particularly affecting cyclooxygenase-1). Aspirin is the prototypic agent.
Any of the physical processes that can cause urticaria (reviewed in more detail in the urticaria synopsis) can also be associated with angioedema. These include cold urticaria, cholinergic urticaria, dermatographism, local heat urticaria, solar urticaria, aquagenic urticaria, and delayed pressure urticaria. While these are all predominately urticarial, there is one exception. Hereditary (or acquired) vibratory angioedema has swelling as the more obvious manifestation. Vibratory stimuli such as hammering or rubbing a towel across the back after showering cause dramatic swelling within a few minutes.
Chronic Spontaneous Urticaria (CSU) is a separate entity in which urticaria persists for at least 6 weeks; it is presumed that symptoms are present most days of the week. The incidence of angioedema with the urticaria is 40% and its presence adversely affects symptom scores and quality of life. The disorder has a strong association with autoimmunity and autoallergy. Autoimmune associations include a 40% incidence of IgG antibody to the alpha subunit of the IgE receptor, a 25% incidence of antithyroid antibodies, and a 30% incidence of positive antinuclear antibody. Anti double-stranded DNA is not seen nor is there an association (clinically) with systemic lupus erythematosus (SLE) even though some patients with SLE have urticaria and/or angioedema. There is an increased incidence of Hashimoto’s Thyroiditis, and to a lesser degree, type I diabetes and alopecia. Autoallergy is a consideration because total IgE is elevated and there is an increased incidence of IgE anti-thyroperioxidase, and a striking increase of IgE anti interleukin 27 that is as high as 75-80%.
Chronic Histaminergic Angioedema
About 15-20% of patients have angioedema that is chronic (i.e. recurrent over long periods of time without identifiable cause) previously designated “idiopathic” angioedema. Many consider it to be the same disorder as chronic spontaneous urticaria (CSU) but without the urticaria. When responsive to high-dose antihistamine therapy, it is now called idiopathic histaminergic angioedema since it is histamine and mast cell dependent. Nevertheless, an alternative consideration is that it might be pathogenically different from CSU because it lacks the female predominance or the high percentage of auto antibodies.
Angioedema without Urticaria
This designation is utilized for entities where angioedema is always seen in the absence of urticaria, whether it is acute or chronic (i.e. recurrent). Most of the disorders in this category are caused by bradykinin and are not associated with mast cell degranulation or release of histamine. They are listed in Table I.

Hereditary C1 Inhibitor Deficiency (Types I and II Hereditary Angioedema)
This hereditary disorder is due to a mutation in the gene for C1 inhibitor (C1 INH). Its incidence is 1:20,000 to 1:50,000 and is autosomal, i.e. affects men and women equally. However the severity and frequency occurrence is associated with trauma, infection, medication containing estrogen, and stress, although at least 50% of episodes have no identifiable precipitant. There are three types of areas of swelling; namely peripheral, gastrointestinal, or laryngeal. The typical appearance of a facial swelling is shown in Fig. 1.
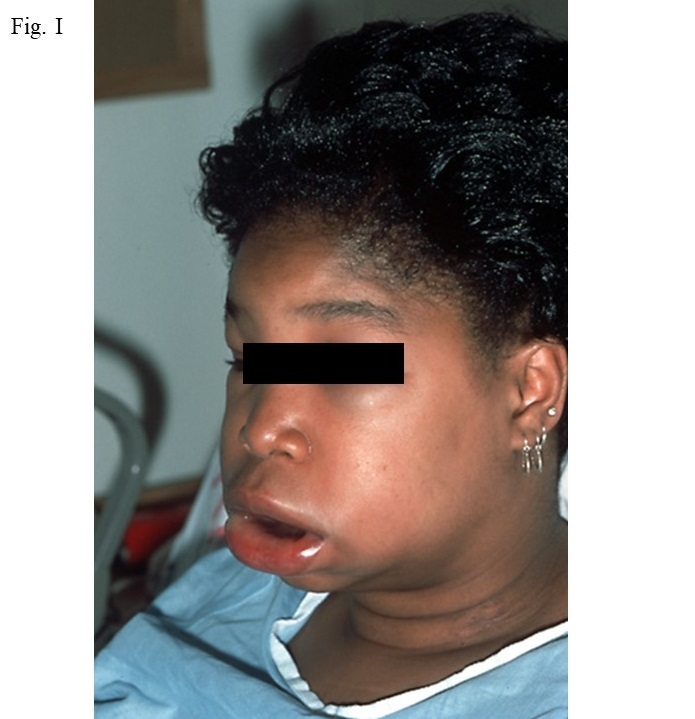
Gastrointestinal swelling is due to edema of the bowel wall with symptoms of severe abdominal pain, vomiting, and/or diarrhea. A typical episode lasts 3 days, often leads to visits to the emergency facility, and can result in surgery that is unnecessary because symptoms are similar to those of an “acute abdomen” or bowel obstruction. Excessive opiate administration can lead to addiction. Laryngeal attacks are potentially fatal so maintenance of an airway by intubation or tracheostomy is essential if stridor becomes evident.
C1 inhibitor deficiency presents as an autosomal dominant disorder so that there is one mutated gene and one normal one. The C1 INH blood level is typically below the expected 50% because of hypermetabolism and/or depressed mRNA transcription. Attacks of angioedema typically occur when blood levels are less than 40% and most patients fall within a range of barely detectable to 35%. Yet 25% of patients do not report a positive family history of angioedema. Most represent new mutations so there is no parenteral or sibling angioedema but their children would have a 50% chance of receiving the mutant gene. C1 inhibitor stabilizes the C1 complex containing C1q, C1r, and C1s. When blood levels are insufficient, C1r autoactivates, converts C1s to activated C1s, which in turn digests C4. As a result the C4 level is low in 95% of patients even at times when they are asymptomatic. During an attack of angioedema, the C4 levels declines further and often approaches zero and C2 (the second substrate of activated C1s) is activated and depleted. C2 is, however, normal when patients’ disease is quiescent. A low C4 in a patient with recurrent angioedema suggests C1 INH deficiency and one would determine the C1 INH level by protein and by function. In type I HAE, synthesis is markedly curtailed so that the quantitative protein level and function are both low, representing about 85% of patients. Type II HAE has a normal protein level but low C1 INH function due to a mutation (usually an amino acid substitution in exon 8) that impedes its inhibitory action.
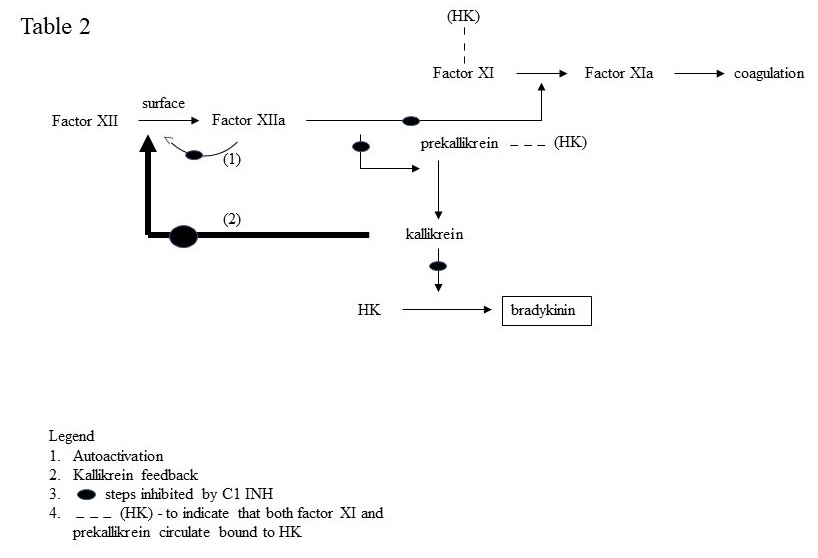
The swelling characteristic of C1 inhibition is caused by over production of bradykinin. C1 INH inhibits two of the plasma enzymes requisite for bradykinin formation; namely, activated factor XII and plasma kallikrein as depicted in Fig. 2.

Factor XII has intrinsic activity estimated to be about 1/40,000 that of activated (cleaved) factor XII. It is slowly autoactivated when bound to negatively charged surfaces. Activated factor XII (factor XIIa or factor XIIf) then converts prekallikrein to kallikrein and kallikrein digests high molecular weight kininogen (HK) to release bradykinin. A positive feedback is critical in which kallikrein rapidly converts factor XII to factor XIIa (80,000 molecular weight) and then to factor XIIf (32,000 molecular weight). Factor XIIa also activates factor XI to continue the intrinsic coagulation cascade. Factor XIIf lacks the surface binding site, loses 96% of the coagulant activity of factor XIIa but continues to activate prekallikrein and gains the ability to convert C1r to activated C1r thereby linking bradykinin formation to further complement activation (not shown). The substrates of activated factor XII (i.e. factor XI and prekallikrein) both circulate bound to HK and HK functions as a cofactor that increases the rate of activation of factor XI and prekallikrein and indirectly enhances factor XII activation (i.e. via kallikrein).
The important functions of C1 INH are shown in Table II.
HAE with Factor XII Mutation
This is one of three described forms of HAE with normal C1 INH. Excess bradykinin formation appears to be due to a rapid cleavage of mutated factor XII by plasmin at the maturation site followed by cleavage of the usual activating bond by kallikrein or plasmin. The net result is an enhanced rate of bradykinin formation that is not controlled by C1 INH even in normal quantities. There is an even more striking association of clinical symptoms with female sex and exposure to any exogenous estrogen. Symptoms are very similar to that seen with C1 inhibitor deficiency.
HAE with Plasminogen Mutation
While HAE-factor XII is linked to fibrinolysis because of the creation of a new plasmin cleavage site, here the mutation is in the precursor plasminogen molecule. The molecular mechanism by which plasminogen function(s) is affected is unknown but overproduction of bradykinin does appear to be the outcome like the other forms of HAE.
HAE with Kininogen Mutation
The first family was recently described with a mutation just preceding the bradykinin sequence. Thus it is present in both high molecular weight kininogen and low molecular weight kininogen and could therefore relate to both plasma kallikrein and/or tissue kallikrein.
HAE with Angiopoietin Mutation
This very rare form of HAE is inherited as an autosomal dominant disorder, as is all of the others, but is not a result of bradykinin overproduction. Rather, angiopoietin is linked to the bradykinin receptor and that of vascular endothelial growth factor and is thought to enhance the function of bradykinin at the B-2 receptor level.
Bradykinin causes angioedema by stimulating contractile elements in endothelial cells so as to create spaces (“pores”) between the cells through which fluid can pass. Secondarly, nitric oxide and prostaglandin formation is enhanced.
Additional forms of HAE i.e. familial recurrent angioedema undoubtedly exist and are considered HAE-unknown. This designation is not appropriate for recurrent angioedema of unclear origin if a clear family history is not evident.
Acquired C1 Inhibitor Deficiency
This disorder is most commonly seen in men and typically becomes evident in the 50’s. It is due to IgG antibody to C1 INH (present in close to 90% of patients) thus it is an autoimmune disorder. The C4 is low, like HAE with C1 INH deficiency and quantitation of C1 INH by protein or function is greatly diminished. Thus it resembles type I HAE. The antibody interferes with the C1 INH quantitation and the C1 INH is dysfunctional, in part due to antibody binding as well as the presence of inactive cleavage fragments. This disorder is associated with monoclonal gammapathy where the monoclonal protein is anti-C1 INH as well as B-cell lymphoma. A protein electrophoresis and careful physical exam is indicated when this entity is suspected. It differs from HAE-C1 INH deficiency in that C1q protein level is diminished in about 70% of patients so a normal value does not eliminate it as a consideration but a low value is highly suggestive. This is likely due to immune complex formation between C1 INH (the antigen) and the antibody. Circulating immune complexes may also be present involving tumor antigens with B-cell lymphoma is present.
Angioedema Due to Inhibition of Angiotensin Converting Enzyme (ACE)
This is the most common form of angioedema seen in emergency rooms and is presumed (but not proven) to be caused by bradykinin accumulation. The clinical presentation is like HAE but has lesser GI involvement and a predilection to involved face, tongue (which can become huge) and larynx. Asphyxiation is an ever-present threat. ACE inhibitors block conversion of angiotensin I to the potent vasoconstrictor angiotensin II and also prevent bradykinin degradation by ACE, hence bradykinin levels can rise even with a normal synthetic rate.
Idiopathic Non-Histaminergic Angioedema
Recurrent angioedema on the absence of any positive family history of swelling that is also unresponsive to antihistamines taken up to 4 times a day falls into this category. It is thought to be caused by bradykinin, however if so, the mechanism is unknown. Case reports suggest responsiveness to agents used to treat other forms of bradykinin-mediated angioedema. In contrast to the histaminergic group, gastrointestinal and laryngeal attacks of swelling can be present.
Treatment of Angioedema
Angioedema Related to Mast Cells
These include acute and chronic urticaria and angioedema, idiopathic histaminergic angioedema, inducible (physical) urticaria, and various forms of allergic and non-allergic angioedemas that are at least particularly responsive to antihistamines, are thought to require mast cell (and possibly basophil) degranulation, and respond to corticosteroid if there is a cellular infiltrate. Many are known to respond to omalizumab, a monoclonal antibody that sequesters IgE.
Antihistamines are used to treat all of these. Often the dose required is four times a day employing newer non-sedating choices. Unresponsiveness may require a short course of corticosteroid or longer-term therapy with omalizumab. Angioedema is self-limited so that individual episodes can be treated with 2-3 days of steroid e.g. 20-40 mg in a single dose and then stop without a taper. Chronic steroid therapy is no longer recommended for any of the above diagnoses.
Inducible urticarias are treated with antihistamine up to 4 times daily. If unsuccessful and because these persist for long periods of time, omalizumab is the next choice. It has been studied in cold urticaria, cholinergic urticaria, and dermatographism, the most common forms with case reports suggesting efficacy in the others. Delayed pressure urticaria is the only inducible urticaria with a substantial cellular infiltrate. It is typically unresponsive to antihistamine and is steroid responsive but requires high dosage. Omalizumab is the drug of choice. For omalizumab failures, cyclosporine can be tried.
Angioedema accompanying chronic spontaneous urticaria is responsive to the approach advocated by guidelines. One employs non-sedating antihistamines up to 4 times a day, and omalizumab for antihistamine failures. While not formally studied beyond the 300 mg/month dose, one can increase to 450 mg or even 600 mg for refractory patients or administer the 300 mg every 2-3 wks.
Idiopathic histaminergic angioedema can be treated acutely with antihistamines, but 2-3 days of corticosteroid is more effective. If frequency is high, one can try prophylaxis with antihistamine taken 4 times a day. Omalizumab is another choice for refractory or frequent events but we lack controlled studies as has been done for CSU.
Angioedema Caused by Bradykinin
Therapy can be divided into approaches that deal with interrupting an acute episode vs those administered prophylactically to prevent attacks of swelling. Numerous studies thru phase 3 have been successfully completed with approvals in countries throughout the world for HAE types I and II.
Acute attacks of swelling can be treated with an intravenous infusion of C1 INH (purified or recombinant), a subcutaneous injection of Icatibant, a bradykinin antagonist that binds to the B-2 receptor, or a subcutaneous injection of Ecallantide, an inhibitor of plasma kallikrein. Prophylactic therapy can be provided by regular administration of C1 INH, typically twice weekly. The best results are obtained with a subcutaneous preparation in terms of efficacy which can achieve high stable C1 INH levels in the 70-80% of normal range. Intravenous preparations are available and have been employed for many years using protein purified from plasma or cloned C1 INH produced in rabbits. Another approach is subcutaneous administration of a monoclonal antibody to plasma kallikrein every 2 weeks. It may be similarly effective as subcutaneous C1 INH. An oral approach employing an inhibitor of plasma kallikrein taken daily has just completed a phase 3 study.
Any of these agents can be employed in the treatment of the multiple types of HAE with normal C1 INH although controlled studies of these relatively rare syndromes have not been reported. The antifibrinolytic agent tranexamic acid may be particularly useful in the treatment of HAE with factor XII mutation since it is particularly susceptible to activation by plasmin. Perhaps it can be tried in those with the mutant plasminogen. Administration of progesterone may also be particularly efficacious in patients with HAE and factor XII mutation because it is so dependent on estrogen.
Patients with acquired C1 INH deficiency require therapy directed to lymphoma if one is present. For those with a monoclonal anti C1 INH, plasmapheresis acutely plus rituximab can be directed to the B cell hyperreactivity. Otherwise attacks of swelling can be treated as one would approach type I HAE with the addition that tranexamic acid does seem to be more effective in the acquired disorder than the hereditary (types I and II) disorder. Tranexamic acid can also be helpful in the prophylactic treatment of children with C1 INH deficiency.
Treatment of idiopathic non-histaminergic angioedema can be approached with the same choices employed for type I HAE.
Treatment of ACE inhibitor angioedema is to avoid all ACE inhibitor preparations. Protecting the airway is the most important point for the acute occurrence. Blocking bradykinin with Icatibant ought to be helpful. The same may be true for HAE-N with angiopoitin 1-mutation. Angiotensin receptor blocking agents (ARB’s) are, in general, well-tolerated in patients who have had angioedema due to an ACE inhibitor.